Advanced Usage
This page collects the controls most users need after the first successful run: levels of theory, job selection, ESS routing, resource control, rotor scans, transition-state adapters, restarts, and troubleshooting.
Flexible Coordinate Input
The xyz field of an ARCSpecies can be:
a multiline XYZ string;
a list of XYZ strings;
a path to an XYZ file;
a path to a supported ESS input or output file;
a path to ARC conformer files generated before or after optimization.
Example:
species:
- label: TS1
is_ts: true
xyz:
- guesses/ts1_guess_1.gjf
- guesses/ts1_guess_2.out
- |
C 0.000000 0.000000 0.000000
H 0.000000 0.000000 1.089000
Job Types
ARC recognizes these current job type keys:
conf_opt- conformer optimization;conf_sp- conformer single-point jobs;opt- geometry optimization;fine- fine-grid optimization;freq- frequency calculation;sp- single-point energy;rotors- rotor scans;irc- intrinsic reaction coordinate;orbitals- molecular orbitals;onedmin- Lennard-Jones / OneDMin workflow;bde- bond dissociation energy workflow.
Older input aliases are still normalized in code: fine_grid maps to
fine and lennard_jones maps to onedmin. Prefer the current names in
new inputs.
Run one job family:
specific_job_type: sp
When specific_job_type is set, it takes precedence over job_types.
Levels of Theory
The fastest way to specify a common workflow is level_of_theory:
level_of_theory: CCSD(T)-F12/cc-pVTZ-F12//wb97xd/def2tzvp
This means:
optimize, frequency, and scan jobs use
wb97xd/def2tzvp;single-point jobs use
CCSD(T)-F12/cc-pVTZ-F12.
A single non-composite method applies to opt, freq, scan, and sp:
level_of_theory: wb97xd/def2svp
A composite method is specified without a slash:
level_of_theory: CBS-QB3
Use job-specific keys when you need more control:
conformer_opt_level:
method: b3lyp
basis: 6-31g(d,p)
dispersion: empiricaldispersion=gd3bj
opt_level: wb97xd/def2tzvp
freq_level: wb97xd/def2tzvp
sp_level:
method: DLPNO-CCSD(T)-F12
basis: cc-pVTZ-F12
auxiliary_basis: aug-cc-pVTZ/C
cabs: cc-pVTZ-F12-CABS
software: orca
Do not put Arkane correction years into QC method names such as
wb97xd32023. Use arkane_level_of_theory.year when you need a specific
Arkane correction year:
arkane_level_of_theory:
method: b97d3
basis: def2tzvp
year: 2023
ESS-Specific Arguments
Use args for extra ESS keywords or blocks:
opt_level:
method: wb97xd
basis: def2tzvp
software: gaussian
args:
keyword:
general: iop(99/33=1)
For multiline blocks:
sp_level:
method: dlpno-ccsd(t)
basis: def2tzvp
software: orca
args:
block:
general: |
%scf
MaxIter 500
end
Multireference Methods (MRCI)
To request a multireference calculation such as MRCI, specify any of the
following on sp_level. A “simple” MRCI computation:
sp_level: MRCI/cc-pVTZ
Explicitly correlated (F12) calculations improve basis-set convergence and are only available through Molpro:
sp_level:
method: MRCI-F12
basis: cc-pVTZ-F12
You can also specify a chain of jobs (supported in Molpro and Orca) so that the MRCI calculation uses the orbitals of the previous job. For example, to perform an MRCI calculation on CASSCF orbitals:
sp_level:
method: MP2_CASSCF_MRCI
basis: aug-cc-pVTZ
This chain, separated by underscores, performs an HF calculation (by default, no
need to specify), an MP2 calculation, then a CASSCF calculation, and finally an
MRCI calculation on the CASSCF orbitals. Requesting an MRCI job causes ARC to first
automatically spawn a Molpro CCSD/cc-pVDZ job to identify the active space for the
MRCI calculation. If the subsequent job is spawned in Orca, the active space is
used; if it is spawned in Molpro, the entire space is currently considered (the
active space is not determined explicitly). It is therefore recommended to set the
levels_ess dict in settings so that MRCI jobs run in Orca, and F12 and
CCSD jobs run in Molpro.
ARC extracts active-space parameters from the Molpro CCSD output file to guide the subsequent calculation:
Active electrons are obtained by subtracting the net charge and the core electrons (estimated as 2 per heavy atom) from the total nuclear charge:
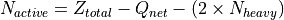
Active orbitals are determined by summing the counts of “closed-shell” and “active” orbitals reported in the output.
The active-space routine returns a dictionary containing the 'e_o' tuple
(electrons, orbitals) alongside lists of occupied ('occ') and closed-shell
('closed') orbitals per irreducible representation.
Solvation
Solvation is specified on a level of theory with the top-level fields
solvation_method and solvent:
opt_level:
method: wb97xd
basis: def2tzvp
software: gaussian
solvation_method: pcm
solvent: diethylether
Support is adapter-dependent. Gaussian, ORCA, and xTB currently have solvation handling in their job adapters; always choose method and solvent names in the format expected by the selected ESS.
Adaptive Levels
Use adaptive_levels to change methods by molecule size. ARC expects tuple
keys for the heavy-atom ranges and tuple keys for grouped job types. In an
input.yml file, write tuple keys with YAML’s !!python/tuple tag:
adaptive_levels:
? !!python/tuple [1, 5]
:
? !!python/tuple [opt, freq]
: wb97xd/6-311+g(2d,2p)
sp: ccsd(t)-f12/aug-cc-pvtz-f12
? !!python/tuple [6, 15]
:
? !!python/tuple [opt, freq]
: b3lyp/cbsb7
sp: dlpno-ccsd(t)/def2-tzvp
? !!python/tuple [16, inf]
:
? !!python/tuple [opt, freq]
: b3lyp/6-31g(d,p)
sp: wb97xd/6-311+g(2d,2p)
When using ARC from Python, pass regular Python tuples:
adaptive_levels = {
(1, 5): {
('opt', 'freq'): 'wb97xd/6-311+g(2d,2p)',
'sp': 'ccsd(t)-f12/aug-cc-pvtz-f12',
},
(6, 15): {
('opt', 'freq'): 'b3lyp/cbsb7',
'sp': 'dlpno-ccsd(t)/def2-tzvp',
},
(16, 'inf'): {
('opt', 'freq'): 'b3lyp/6-31g(d,p)',
'sp': 'wb97xd/6-311+g(2d,2p)',
},
}
Cover the full heavy-atom range without gaps.
Memory, CPUs, and Wall Time
Set defaults per project:
job_memory: 32
max_job_time: 48
Server entries can also define node limits:
servers = {
'my_slurm': {
'cluster_soft': 'Slurm',
'address': 'login.cluster.edu',
'un': 'my_user',
'key': '/home/my_user/.ssh/id_rsa',
'cpus': 32,
'memory': 128,
},
}
ARC may increase resources during troubleshooting, bounded by server and default job settings. By default, troubleshooting will not request more than 95% of a server node’s configured memory.
Project Directories
By default, command-line runs use the directory containing the input file as the
project directory, while API runs create projects under ARC/Projects. Set
project_directory when you want outputs elsewhere:
project: ethanol_thermo
project_directory: /scratch/my_user/arc_projects/ethanol_thermo
Remote project files are created on the server selected for each job. If a server
entry defines path, ARC uses that path as the base for remote project
storage.
Routing ESS Jobs
Use ess_settings to override global software routing for a project:
ess_settings:
gaussian:
- high_memory_cluster
- local
orca: local
molpro: server2
The order matters when a list is supplied; ARC tries the listed servers in priority order.
Current supported ESS keys include cfour, gaussian, mockter,
molpro, orca, qchem, terachem, onedmin, xtb,
torchani, and openbabel. Some additional adapters, such as TS-search
adapters, are configured through their own settings.
Fine-Grid Optimizations
The fine job type is enabled by default. If fine is true and opt is
false, ARC still runs optimization jobs but treats them as fine-grid jobs from
the start.
job_types:
opt: false
fine: true
Although this argument is called fine in ARC, in practice it directs the ESS to
use an ultrafine grid. See, for example, this study describing the
importance of the DFT grid.
In Gaussian, fine adds the following directive:
scf=(tight, direct) integral=(grid=ultrafine, Acc2E=12)
In QChem, it adds the following directives:
GEOM_OPT_TOL_GRADIENT 15
GEOM_OPT_TOL_DISPLACEMENT 60
GEOM_OPT_TOL_ENERGY 5
XC_GRID 3
In TeraChem, it adds the following directives:
dftgrid 4
dynamicgrid yes
Rotor Scans
rotors is enabled by default. ARC identifies internal rotors and runs scans
for valid torsions. The default scan resolution is controlled by
rotor_scan_resolution in settings.
Disable rotor scans for a project:
job_types:
rotors: false
Use directed_rotors or preserve_param_in_scan on species when you need
more control over scan definitions and constrained internal coordinates.
ND Rotor Scans
ARC also supports ND (N-dimensional, N >= 1) rotor scans. There are seven different ND types to execute:
A1. Generate all geometries in advance (brute force), and calculate single-point energies (nested or diagonalized).
A2. Generate all geometries in advance (brute force), and run constraint optimizations (nested or diagonalized).
B. Derive the geometry from the previous point (continuous) and run constraint optimizations (nested or diagonalized).
Let the ESS guide the optimizations.
Each of the options above (A or B) can be either “nested” (considering all ND dihedral combinations) or “diagonal” (resulting in a unique 1D rotor scan across several dimensions). The seventh option (C) allows the ESS to control the ND scan, which is similar in principle to option B, but not directly controlled by ARC.
The optional primary keys are:
brute_force_spbrute_force_optcont_optess
The brute-force methods generate all the geometries in advance and submit all relevant jobs simultaneously. The continuous method waits for the previous job to terminate, and uses its geometry as the initial guess for the next job.
Another set of three keys is allowed, adding _diagonal to each of the above keys.
The secondary keys are therefore:
brute_force_sp_diagonalbrute_force_opt_diagonalcont_opt_diagonal
Specifying _diagonal increments all the respective dihedrals together, resulting
in a 1D scan instead of an ND scan. Values are nested lists. Each value is a list
where the entries are either pivot lists (e.g., [1, 5]) or lists of pivot lists
(e.g., [[1, 5], [6, 8]]), or a mix (e.g., [[4, 8], [[6, 9], [3, 4]]]). The
requested directed scan type is executed separately for each list entry. A list entry
that contains only two pivots results in a 1D scan, while a list entry with N pivots
considers all of them and results in an ND scan (if _diagonal is not specified).
Note that indices are 1-indexed.
ARC generates geometries using the rotor_scan_resolution argument in
settings.py. An 'all' string entry is also allowed in the value list,
triggering a directed internal-rotation scan for all torsions in the molecule. If
'all' is specified within a second-level list, all the dihedrals are considered
together. Currently ARC does not automatically identify torsions to be treated as ND,
so this attribute must be specified by the user.
To execute ND rotor scans, first set the rotors job type to True, then set
the directed_rotors attribute of the relevant species. Below are several examples.
To run all dihedral scans of a species separately using brute-force sp (each as 1D):
spc1 = ARCSpecies(label='some_label', smiles='species_smiles', directed_rotors={'brute_force_sp': ['all']})
To run all dihedral scans of a species as a conjugated scan (ND, N = the number of torsions):
spc1 = ARCSpecies(label='some_label', smiles='species_smiles', directed_rotors={'cont_opt': [['all']]})
Note the change in list level (all is either within one or two nested lists) in
the above examples.
To run specific dihedrals as ND (here all 2D combinations for a species with 3 torsions):
spc1 = ARCSpecies(label='C4O2', smiles='[O]CCCC=O', xyz=xyz,
directed_rotors={'brute_force_opt': [[[5, 3], [3, 4]], [[3, 4], [4, 6]], [[5, 3], [4, 6]]]})
Note: ND rotors are still not incorporated into the molecular partition function, so they currently do not affect thermo or rates.
Note: Any torsion defined as part of an ND rotor scan will not be spawned for that species as a separate 1D scan.
Warning: Job arrays have not been incorporated into ARC yet. Spawning ND rotor scans will result in many individual jobs being submitted to your server queue system.
Transition-State Search Adapters
ARC can use several TS adapters when configured and installed, including heuristics, linear, AutoTST, KinBot, GCN, xTB-GSM, and ORCA-NEB. See Transition State Search for a description of each. Select adapters per project:
ts_adapters:
- heuristics
- xtb_gsm
- orca_neb
User-supplied ts_xyz_guess values are always a useful fallback because they
make the calculation less dependent on automated TS guess generation.
Pipe Mode
Pipe mode is ARC’s opt-in distributed execution path for large homogeneous job batches on HPC systems. It is disabled by default:
pipe_settings = {
'enabled': True,
'min_tasks': 10,
'lease_duration_hrs': 1,
}
Enable it in ~/.arc/settings.py only after your normal scheduler submission
works. ARC considers pipe mode for eligible batches once min_tasks is met.
Transition-state guess generation is not currently wired through pipe mode, so
do not rely on pipe mode for TS-guess orchestration.
Troubleshooting Controls
ARC attempts ESS and rotor troubleshooting by default. Disable these only when you need strict no-resubmission behavior:
trsh_ess_jobs: false
trsh_rotors: false
Use keep_checks: true when Gaussian checkfiles or other retained files are
needed for manual diagnosis.
At times a user might know in advance that a particular additional keyword is
required for the calculation. In such cases, pass the relevant keyword in the
initial_trsh dictionary (trsh stands for troubleshooting), keyed by ESS:
initial_trsh:
gaussian:
- iop(1/18=1)
molpro:
- shift,-1.0,-0.5;
qchem:
- GEOM_OPT_MAX_CYCLES 250
Batch Delete ARC Jobs
Warning
DANGER ZONE: make sure you understand what you’re doing before running this script. Data of running jobs will be lost.
ARC has a feature that deletes all ARC-spawned jobs from selected servers and
projects. To delete all ARC jobs, run the following in the ARC code folder after
activating arc_env:
python arc/utils/delete.py -a
You can also delete jobs from a specific server by specifying its name after the
-s flag:
python arc/utils/delete.py -s server1 -a
To delete jobs from a specific ARC project, pass the project’s name after the
-p flag:
python arc/utils/delete.py -p project1
Alternatively (since project names might be long and not always shown in full when requesting the server job status), you can supply an ARC job ID, and ALL jobs related to the project of the given job ID will be deleted (NOT only the given job!):
python arc/utils/delete.py -j a_54836
Note that either a -a, a -p, or a -j flag must be given. All flags can
be combined with the optional -s flag.
Writing an ARC Input File Using the API
Writing YAML by hand isn’t very intuitive for many users. You can instead use ARC’s API to define your objects, then dump them into a YAML file that ARC can read as an input:
from arc.species.species import ARCSpecies
from arc.common import save_yaml_file
input_dict = dict()
input_dict['project'] = 'Demo_project_input_file_from_API'
input_dict['job_types'] = {'conf_opt': True,
'opt': True,
'fine': True,
'freq': True,
'sp': True,
'rotors': True,
'conf_sp': False,
'orbitals': False,
'lennard_jones': False,
}
spc1 = ARCSpecies(label='NO', smiles='[N]=O')
adj1 = """multiplicity 2
1 C u0 p0 c0 {2,D} {4,S} {5,S}
2 C u0 p0 c0 {1,D} {3,S} {6,S}
3 O u1 p2 c0 {2,S}
4 H u0 p0 c0 {1,S}
5 H u0 p0 c0 {1,S}
6 H u0 p0 c0 {2,S}"""
xyz2 = [
"""O 1.35170118 -1.00275231 -0.48283333
C -0.67437022 0.01989281 0.16029161
C 0.62797113 -0.03193934 -0.15151370
H -1.14812497 0.95492850 0.42742905
H -1.27300665 -0.88397696 0.14797321
H 1.11582953 0.94384729 -0.10134685""",
"""O 1.49847909 -0.87864716 0.21971764
C -0.69134542 -0.01812252 0.05076812
C 0.64534929 0.00412787 -0.04279617
H -1.19713983 -0.90988817 0.40350584
H -1.28488154 0.84437992 -0.22108130
H 1.02953840 0.95815005 -0.41011413"""]
spc2 = ARCSpecies(label='vinoxy', xyz=xyz2, adjlist=adj1)
spc_list = [spc1, spc2]
input_dict['species'] = [spc.as_dict() for spc in spc_list]
save_yaml_file(path='some/path/to/desired/folder/input.yml', content=input_dict)
The above code generates the following input file:
project: Demo_project_input_file_from_API
job_types:
rotors: true
conf_opt: true
fine: true
freq: true
lennard_jones: false
opt: true
orbitals: false
sp: true
species:
- E0: null
arkane_file: null
bond_corrections:
N=O: 1
charge: 0
external_symmetry: null
force_field: MMFF94
generate_thermo: true
is_ts: false
label: 'NO'
mol: |
multiplicity 2
1 N u1 p1 c0 {2,D}
2 O u0 p2 c0 {1,D}
multiplicity: 2
number_of_rotors: 0
- E0: null
arkane_file: null
bond_corrections:
C-H: 3
C-O: 1
C=C: 1
charge: 0
conformers:
- |-
O 1.35170118 -1.00275231 -0.48283333
C -0.67437022 0.01989281 0.16029161
C 0.62797113 -0.03193934 -0.15151370
H -1.14812497 0.95492850 0.42742905
H -1.27300665 -0.88397696 0.14797321
H 1.11582953 0.94384729 -0.10134685
- |-
O 1.49847909 -0.87864716 0.21971764
C -0.69134542 -0.01812252 0.05076812
C 0.64534929 0.00412787 -0.04279617
H -1.19713983 -0.90988817 0.40350584
H -1.28488154 0.84437992 -0.22108130
H 1.02953840 0.95815005 -0.41011413
force_field: MMFF94
generate_thermo: true
is_ts: false
label: vinoxy
mol: |
multiplicity 2
1 O u1 p2 c0 {3,S}
2 C u0 p0 c0 {3,D} {4,S} {5,S}
3 C u0 p0 c0 {1,S} {2,D} {6,S}
4 H u0 p0 c0 {2,S}
5 H u0 p0 c0 {2,S}
6 H u0 p0 c0 {3,S}
multiplicity: 2
number_of_rotors: 0
Restarts
Restart files are normal ARC inputs with more state. To restart:
conda activate arc_env
python /path/to/ARC/ARC.py restart.yml
Keep the project directory and server-side job files available when restarting; ARC uses them to collect and continue previously submitted work.